
搜索结果: 1-15 共查到“化学 电子结构”相关记录153条 . 查询时间(0.74 秒)

电解水被认为是一种可持续的、极具发展前景的产H2策略。通常,电解水通过两个半反应进行:析氧反应(OER)和析氢反应(HER)。实际应用中HER和OER催化剂的有效耦合是非常必要的。尽管已报道了许多耦合催化剂,但仍有一些关键问题需要进一步解决:1)阳极和阴极的催化剂通常在成分上存在很大差异,使制备过程复杂,且由于在电解水过程中可能发生催化剂溶解、再沉积反应,会引起阴极和阳极催化剂相互干扰;2)大部分...
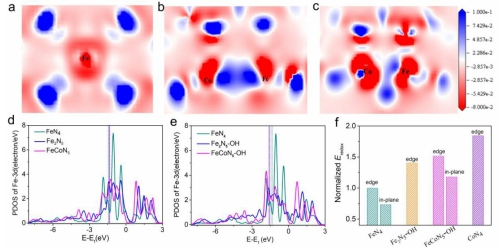
基于此,中国科学院长春应用化学研究所邢巍、葛君杰研究员联合武汉大学陈胜利教授与上海光源姜政研究员设计了FeCoN5双原子位点,水在该中心上自发解离生成新的FeCoN5OH稳定位点,可调控Fe的d-轨道能级,提升Fe的Eredox。结合DFT理论计算与原位X射线近边吸收光谱(XANES)证实该新型活性位点上的Fe(III)/Fe(II)氧化还原电势得到提高,从而大幅提升ORR性能,促进Fe基催化向火...
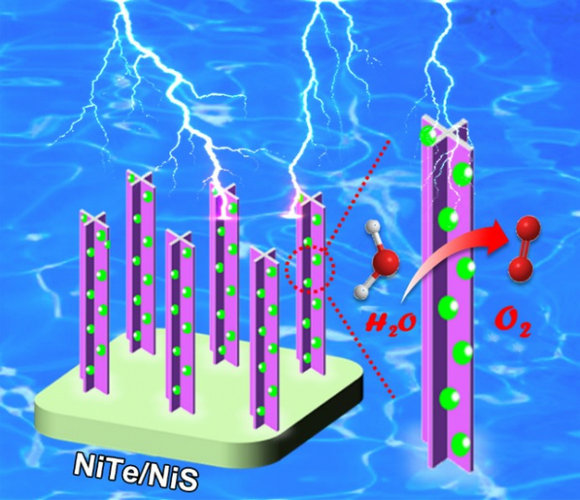
近日,我校化学学院李光琴研究团队通过构建有效的纳米界面来调控催化剂的电子结构,设计制备出高效廉价的氧析出反应异质结电催化剂,并结合实验结果和理论计算对界面调控催化性质的机制进行了研究。他们首先以泡沫镍为基底,通过直接化学刻蚀的方法制备出泡沫镍负载的“十字柱”型的NiTe纳米阵列。进一步通过离子交换反应在NiTe表面修饰NiS纳米颗粒来调控NiTe的电子结构, 制备出NiTe/NiS 异质结催化剂。...

近日,中国科学院大连化学物理研究所分子反应动力学国家重点实验室研究员杨学明、博士周传耀和博士生王志强等与北京计算科学研究中心研究员刘利民以及普林斯顿大学教授Annabella Selloni合作,结合双光子光电子能谱(two-photon photoemission spectroscopy; 2PPE)和理论计算,揭示了二氧化钛中Ti3+离子3d轨道由于John-Teller效应分裂成费米能级以...
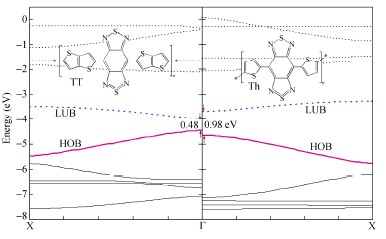
以苯并[1,2-c:4,5-c']二[1,2,5]噻重氮和吡嗪并[2,3-g]喹喔啉为电子受体(A),噻吩、噻吩并[3,2-b]噻吩和二噻吩并[2,3-b:2',3'-d]噻吩为电子供体(D),设计了6种D-A型共轭聚合物.采用B3LYP方法,研究了这6种聚合物的几何结构和电子性质.D-A型共轭聚合物的几何结构和电子结构与电子供体和电子受体的性质,特别是与其提供电子和接受电子的能力密切相关.聚合物...
为了更可靠地得到水溶液中蛋白质分子的电子结构,有必要构建水溶液对蛋白质分子电子结构的等效势,这个等效势必须简单、易用.通过第一性原理、全电子、从头计算,构造了水溶液对酪氨酸(Tyr)电子结构的等效势.工作分为三步:(1)用“自由团簇计算法”计算一个含酪氨酸和水分子的系统的能量最低时的空间结构;(2)基于第一步的空间结构,用“团簇埋入自洽计算法”计算酪氨酸在以水分子为外势条件下的电子结构;(3)用“...
具有18电子结构的Mg2CoH5纳米晶制备及其储氢性能研究
Mg2CoH5纳米晶 储氢性能 制备 球磨法
2012/5/4
本文研究了Mg2CoH5纳米晶的制备及其储氢性能。在室温和氩气气氛下,以MgH2和纳米Co为原料,采用球磨法制备了Mg2CoH5纳米晶。对所制备Mg2CoH5的组成、结构和形貌进行了表征,并且对Mg2CoH5的储氢性能进行了研究。实验结果表明,通过该种方法制备了纯度较高(产物纯度为79%)的四方结构Mg2CoH5纳米晶,其形貌呈球形且分布较均匀,最频粒径为80 nm。制备的Mg2CoH5纳米晶具有...
14族杂原子取代的杂环戊二烯分子具有独特的光谱性质, 成为发光材料的明星分子. 为了更深层次地理解硅、锗、锡杂环戊二烯分子的光谱性质, 本文从理论上计算了它们的电子结构及其吸收和发射光谱. 分别采用密度泛函理论(DFT)和含时密度泛函理论(TD-DFT), 优化了硅、锗、锡杂环戊二烯分子基态和第一激发态的平衡构型, 计算了电子结构和振动性质. 在此基础上, 运用振动关联函数公式计算了吸收光谱和发射...
空位缺陷黄铁矿的电子结构及其浮选行为
电子结构 空位 黄铁矿 浮选
2010/3/9
采用第一性原理平面波赝势计算了含有空位缺陷的黄铁矿的电子结构, 同时讨论了空位缺陷对黄铁矿浮选行为的影响. 研究结果表明, 硫空位对晶胞体积影响不大, 铁空位使黄铁矿晶胞膨胀了1.29%. 空位缺陷主要影响黄铁矿费米能级附近的电子能带结构, 并在禁带中出现了新能级. 空位的存在使黄铁矿的费米能级升高, 不利于黄铁矿的浮选. 另外, 有效质量计算表明, 空位的存在增强了黄铁矿导带底电子的定域性. M...
Sn1-xSbxO2固溶体电极的形成能与电子结构
电子结构 Sb掺杂SnO2 第一性原理 形成能
2010/3/9
为研究Sb掺杂对Ti/SnO2电极稳定性与导电性的影响, 采用基于密度泛函理论的平面波赝势方法对金红石型SnO2及不同比例Sb掺杂SnO2体系进行了第一性原理计算, 用广义梯度近似方法优化了Sn1-xSbxO2固溶体电极的晶体结构, 计算了掺杂前后体系的电子结构以及不同掺杂比例时的形成能. 结果表明: Sb替代Sn后, 晶格常数与晶胞体积均增加, 但掺杂形成能随掺杂量变化不大, 在掺杂量为0.08...
采用密度泛函理论及赝势平面波方法, 对未掺杂SnO2以及过渡金属V、Cr、Mn掺杂SnO2的超原胞体系进行了几何优化, 计算了晶格常数、电子结构与磁学性质. 结果表明, 6.25%与12.5%两种掺杂浓度时, 体系的电子自旋和磁学性质没有发生很大的变化; 相对于未掺杂SnO2, 过渡金属掺杂后SnO2中O原子有向过渡金属移动的趋势, 并使得O与掺杂金属之间键长变短; 在V和Cr掺杂后, SnO2具...
利用密度泛函理论(DFT)中的B3LYP方法优化了氮化钌和氮化锇配合物[M(N)X2]-[M=Ru, Os; X=S2C6H4, mnt(maleonitriledithiolate)]的基态几何结构, 得到的几何参数与实验结果吻合得很好. 采用TD-DFT方法, 得到了配合物在CH3CN溶液中的激发态电子结构和电子吸收光谱. 利用SCRF方法中的CPCM模型来模拟溶剂化效应. 研究结果表明, 配...
分别采用B3LYP/6-31G(d)和CIS/6-31G(d)方法对4-(1,2-二苯基)乙烯基-4’-(N,N-二苯基-4-乙烯基苯胺基)联苯(A)及其二氟取代衍生物(B-F)的基态(S0)和单重激发态(S1)的几何构型进行了全优化, 计算获得了电离势IP、电子亲和势EA等相关数据, 并采用含时密度泛函(TD-DFT)方法计算了上述化合物的电子吸收和荧光发射光谱. 研究结果表明, 化合物A及二氟...
基于广义梯度近似(GGA)的密度泛函理论(DFT), 通过构造铁磁(FM), 阻挫的三角非共线反铁磁(FAFM)、上上下下型共线反铁磁(↑↑↓↓AFM)三种不同磁性构型, 从非共线磁性结构计算出发, 优化了低温铜铁矿CuFeO2晶体材料的几何结构, 研究了磁性结构对电子结构、能隙和磁矩等的作用. 计算发现上上下下型反铁磁自旋排列能促进能隙形成, 总能降低, 磁矩增大. 由于上上下下型反铁磁与阻挫三...
采用全势线性缀加平面波(FPLAPW)方法, 在广义梯度近似(GGA)+自旋轨道耦合(SOC)+自旋极化(SP)下计算了具有AuCu3构型的Pu3M和PuM3 (M=Ga, In, Sn, Ge)化合物的平衡结构、电子结构和形成热. 计算的晶格常数与实验值符合得很好; 态密度分析表明Pu 和M 原子轨道间的杂化作用决定于Pu 6d-Pu 5f、Mp-Pu 6d和Msp-M sp轨道杂化之间的竞争,...